
Alzheimer’s Disease, the Renin-Angiotensin System, and COVID-19
ACE2 at the Crossroads of Neurodegeneration and Viral Infection

I. Alzheimer’s Disease: More Than Amyloid
Alzheimer's disease has long been framed as a proteinopathy defined by extracellular β-amyloid plaques and intracellular tau tangles. While these remain central pathological hallmarks, the explanatory model of the disease has expanded considerably over the past two decades.
Alzheimer’s is now increasingly understood as:
- a disorder of the neurovascular unit,
- a disease involving chronic endothelial dysfunction,
- a condition marked by blood-brain barrier disruption,
- and a state of sustained neuroinflammation.
Cerebral hypoperfusion and microvascular abnormalities often precede overt cognitive decline. Endothelial reactivity becomes impaired. Oxidative stress increases. Microglial activation becomes self-perpetuating.
In this broader framework, vascular and inflammatory regulation are no longer peripheral - they are central.
This is where the renin-angiotensin system enters the discussion.
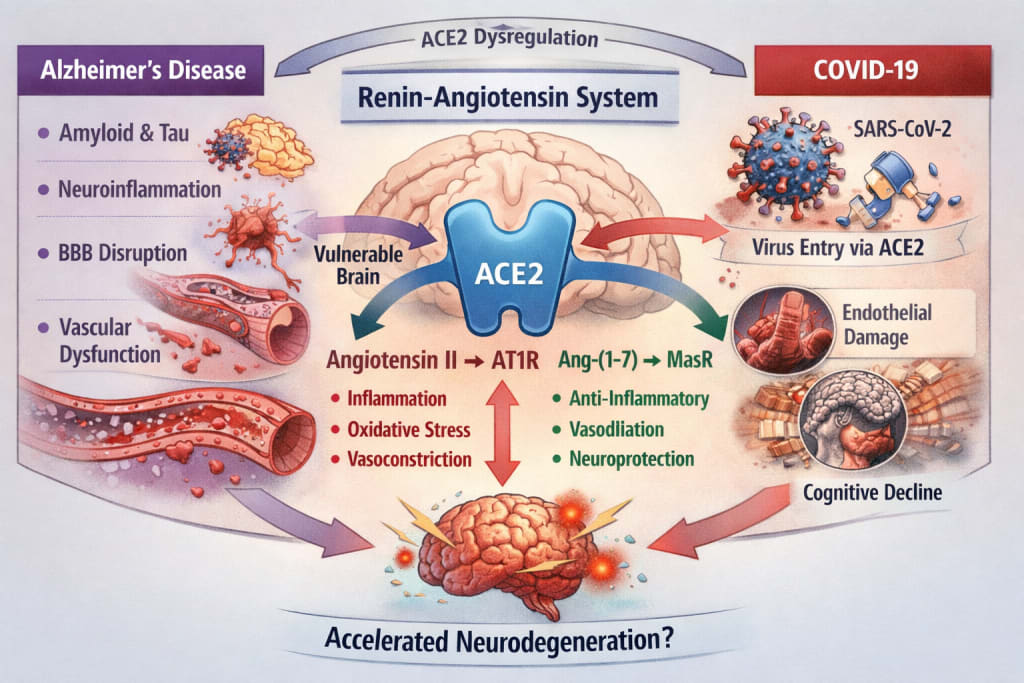
II. The Brain Renin-Angiotensin System: A Delicate Balance
The renin-angiotensin system (RAS) is commonly associated with blood pressure regulation. However, a local RAS exists within the central nervous system, expressed in neurons, astrocytes, microglia, and cerebral endothelial cells.
Its function depends on the balance between two opposing pathways:
The classical axis
- Angiotensin II → AT1 receptor
→ vasoconstriction
→ oxidative stress
→ pro-inflammatory signaling
The counter-regulatory axis
ACE2 → Angiotensin-(1-7) → Mas receptor
→ vasodilation
→ anti-inflammatory effects
→ endothelial protection
Evidence suggests that in Alzheimer’s disease:
- ACE2 expression may be reduced,
- the Ang II / AT1 pathway is relatively overactive,
- oxidative stress and inflammatory cascades are amplified.
Thus, RAS imbalance may contribute to the neurovascular vulnerability observed in Alzheimer’s pathology.
This biological framework becomes particularly relevant when considering SARS-CoV-2.
III. COVID-19 and ACE2: A System Under Acute Stress
The virus responsible for COVID-19, SARS-CoV-2, uses ACE2 as its cellular entry receptor.
Binding of the viral spike protein to ACE2 leads to:
- receptor internalization,
- reduced membrane ACE2 availability,
- relative predominance of angiotensin II signaling.
The downstream effects are well documented in severe COVID-19:
- systemic inflammation,
- endothelial dysfunction,
- prothrombotic states,
- microvascular injury.
The brain - highly dependent on tight autoregulation and endothelial integrity - is not spared.
Neuropathological and clinical observations in severe COVID-19 include:
- blood–brain barrier disruption,
- microglial activation,
- microvascular lesions,
- persistent cognitive impairment in some patients.
The central question, therefore, is not whether COVID-19 directly “causes” Alzheimer’s disease.
It is whether acute RAS dysregulation in a vulnerable brain can accelerate an ongoing neurodegenerative process.
IV. Pathophysiological Convergence
Several mechanisms converge:
- Alzheimer’s disease involves pre-existing neurovascular fragility.
- Brain RAS balance is already shifted toward pro-inflammatory signaling.
- SARS-CoV-2 infection acutely reduces ACE2 activity.
- Systemic inflammation amplifies neuroinflammatory cascades.
- Hypoxia, delirium, and critical illness are independently associated with long-term cognitive decline.
In elderly patients with limited cognitive reserve, severe infection may function as a "second hit."
Rather than initiating Alzheimer’s disease de novo, COVID-19 may plausibly accelerate clinical expression in individuals with preclinical or early pathological changes.
V. Epidemiological Signals - and Their Limits
Large post-pandemic cohort analyses suggest:
- increased incidence of cognitive impairment following severe COVID-19,
- higher risk among hospitalized and critically ill patients,
- difficulty distinguishing Alzheimer’s disease from vascular dementia or post-ICU cognitive syndrome.
Biomarker-driven studies remain limited.
Causality has not been definitively established.
At present, the data support:
- mechanistic plausibility,
- suggestive epidemiological associations,
- but not conclusive proof of direct causation.
Scientific caution is essential.
VI. Broader Implications
The pandemic has highlighted how deeply interconnected vascular biology, immune signaling, and neurodegeneration may be.
ACE2 sits at the intersection of:
- vascular homeostasis,
- inflammatory regulation,
- viral pathogenesis,
- and neurodegenerative vulnerability.
Understanding this intersection may have implications beyond COVID-19:
- the role of RAS-modulating drugs in neuroprotection,
- long-term cognitive surveillance after severe systemic inflammation,
- and a more integrated view of Alzheimer’s as a systemic-vascular disorder rather than solely a protein aggregation disease.
Conclusion
Alzheimer’s disease is no longer viewed solely through the lens of amyloid and tau.
The renin-angiotensin system contributes to cerebral vascular regulation and inflammatory balance.
COVID-19, through ACE2 disruption, perturbs this system acutely.
Whether SARS-CoV-2 infection accelerates neurodegeneration remains under investigation.
But the convergence of these mechanisms suggests that the brain’s vascular-inflammation axis deserves renewed attention in the post-pandemic era.
About the Creator
Alain SUPPINI
I’m Alain — a French critical care anesthesiologist who writes to keep memory alive. Between past and present, medicine and words, I search for what endures.
Keep reading
More stories from Alain SUPPINI and writers in Futurism and other communities.
Mastering French Without Losing Your Sanity: A Realistic Guide for Beginners
Learning French can feel like solving a Rubik’s Cube blindfolded. The tricky pronunciation, the never-ending grammar rules, and the surprising exceptions make it seem impossible. But don’t worry—equipping yourself with key expressions and effective strategies can turn frustration into fluency. Let’s simplify French and make learning enjoyable.
By Alain SUPPINIExclusive • 12 months ago
How Long-Term Forecasts are Formed in the Cryptocurrency Market?
The cryptocurrency market has matured into a complex financial environment where long-term forecasting requires more than short-term price trends. Early market behavior was often driven by speculation and hype, but today’s digital asset ecosystem is shaped by infrastructure development, real-world adoption, regulatory clarity, and macroeconomic conditions.
By Muhammad Irfan Afzal7 days ago in Futurism
Wireless Audio Devices Market: Smart Headphones, Noise Cancellation & Market Forecast
According to IMARC Group's latest research publication, the global wireless audio devices market size reached USD 60.9 Billion in 2024. Looking forward, IMARC Group expects the market to reach USD 204.8 Billion by 2033, exhibiting a growth rate (CAGR) of 14.4% during 2025-2033.
By sujeet. imarcgroup5 days ago in Futurism
Where there's Art there's Heart
Here's a stupid thing: I adore art, but I start to panic whenever I step into a gallery. In the one place I should be at my contented best - surrounded by walls teeming with creative expression - I fall apart. What ought to be an enriching experience, tacitly designed to facilitate the exploration of human empathy and perspective, is for me an overwhelming purgatory of anxiety that compresses me to the point I cannot breathe. At the same time, I experience a sense of extraction, as though my head is being prized open to create a hole so big my sanity could evaporate. Somewhere between these two opposing forces of vice and vortex, I feel myself dissolving in a stream of panic that makes me want to cry; and I feel so daft feeling this way, that all I want to do is run for the hills.
By Caroline Jane4 days ago in Humans




Comments
There are no comments for this story
Be the first to respond and start the conversation.